新一代蛋白质基座模型“书生·Amix”发布,推动AI原生理解与设计蛋白质 最近,生命科学AI
最近,生命科学AI领域迎来一项重要进展。上海人工智能实验室联合清华大学智能产业研究院、复旦大学等顶尖机构,正式推出了新一代蛋白质基座模型——“书生·Amix”。这个新模型在蛋白质理解与设计等一系列核心任务上,都展现出了业界领先的水平。
免费影视、动漫、音乐、游戏、小说资源长期稳定更新! 👉 点此立即查看 👈
此次发布的“书生·Amix”,是在2025年7月其前代模型Amix-1基础上的重大升级。其核心突破在于引入了文本模态,并以创新的扩散大语言模型为核心架构。这意味着,模型能够对自然语言、蛋白质序列、蛋白语义理解和功能序列设计进行统一建模。简单来说,它成功地将蛋白质这一特殊模态,原生地融入了大模型的多模态认知框架。这标志着相关技术正从简单的工具调用,向着对蛋白质进行深度理解、乃至自主原生设计的方向升级。为了更科学地衡量这一进步,研发团队还同步构建了一个名为ProteinArena的评测体系,旨在为蛋白质模型提供公平且严谨的评估标准。
值得关注的是,上海AI实验室宣布将对“书生·Amix”模型及其代码进行全面开源。这一举措旨在为生命科学基础研究提供一个开放、可复现的强大基座,从而加速药物设计、酶工程、合成生物学等关键领域的产业落地进程。
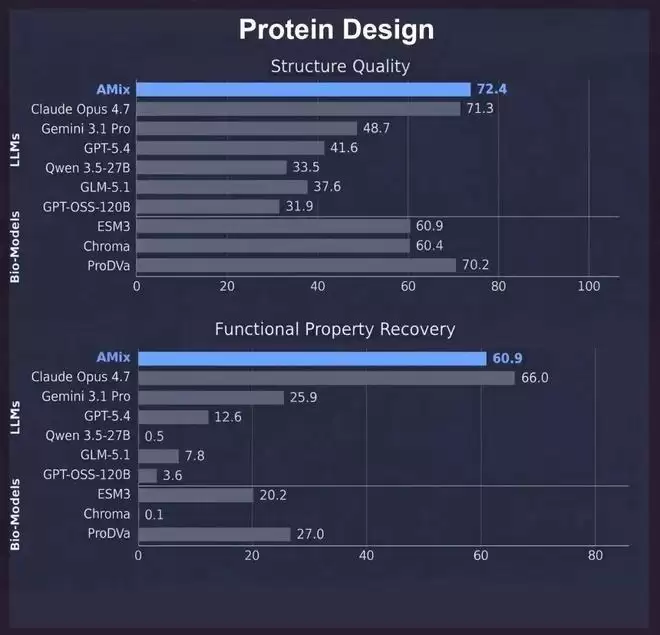
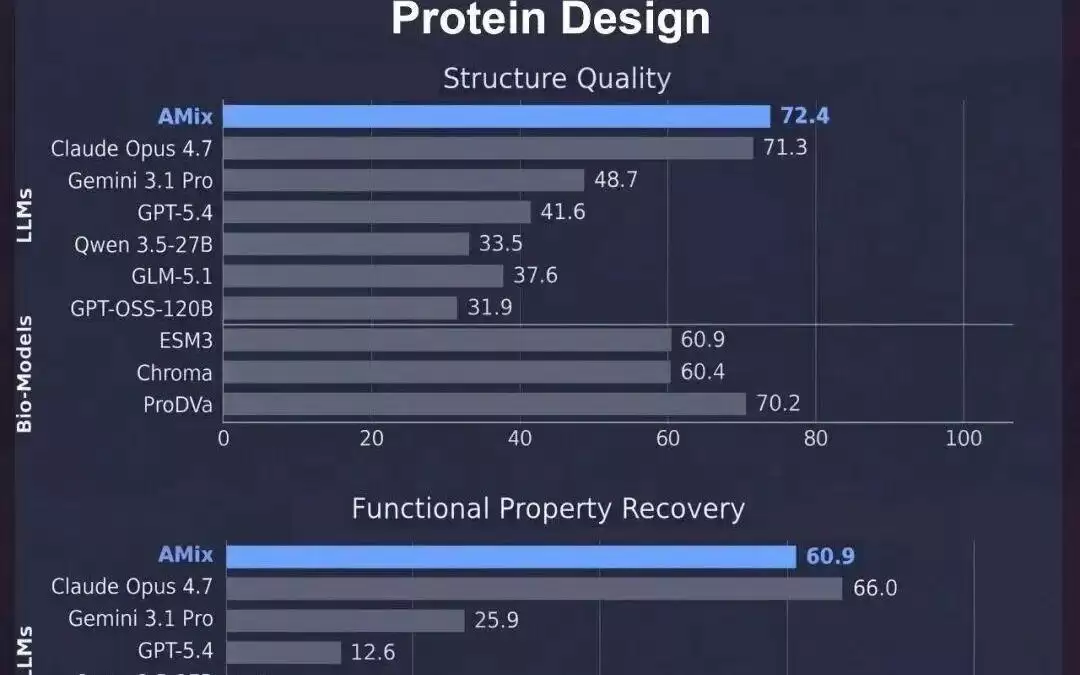
与主流大模型、蛋白质专有模型和工具对比,书生amix在蛋白质理解与设计任务上均表现领先
传统的蛋白质基座模型通常只专注于建模蛋白质序列本身,导致功能呈现“碎片化”特征:用于理解蛋白质功能的模型,和用于设计新序列的模型往往是相互独立的。即便是针对同类任务的细微调整,也需要对模型进行重新训练和微调,效率不高且兼容性欠佳。
“书生·Amix”的突破在于,它打通了自然语言与蛋白质序列之间的壁垒。借助大模型强大的文本指令遵循能力,它实现了蛋白质理解与生成任务的统一。用户只需输入自然语言指令和蛋白质序列,模型就能完成对蛋白质各项功能与性质的解析;而当输入的是蛋白质功能设计指令时,模型则可以直接生成符合要求的蛋白质序列,无需任何额外的模型调试。
实现这一突破的关键,在于蛋白质文本数据的合成与对齐。研究团队整合了Swiss-Prot、InterPro、TremBL等多个权威蛋白质数据库,将其中零散的结构化数据,重新构建成连贯的蛋白质背景知识与指令问答样本。通过联合训练,模型能够将在理解任务中积累的序列与功能关联知识,有效地迁移到生成任务中。这就为蛋白质设计提供了更合理的生物学约束,真正实现了理解与生成能力的双向赋能与协同提升。
在技术架构上,“书生·Amix”采用了扩散大语言模型作为核心。这种通过迭代去噪进行生成的方式,让模型能够同时感知蛋白质的全局功能约束和局部的位点细节。
该架构具备三大显著优势:首先,它天然支持双向上下文理解,能全面兼顾蛋白质序列的前后关联关系;其次,它支持对局部区域进行编辑,便于针对蛋白质的关键功能区域进行精准优化;最后,它能够实现条件可控生成,即依据具体需求生成符合约束条件的蛋白质。在数据量少、约束条件多、组合依赖高的蛋白质研究场景中,扩散语言模型相比传统的自回归模型,展现出更高的数据效率。它突破了单向解码的监督信号局限,通过学习“在任意条件与掩码下的序列补全”,表现出了更优的泛化能力。
这里需要特别指出的是,“书生·Amix”采用了块级扩散范式。其核心机制可以概括为“块间因果约束、块内双向扩散”:在数据块之间,它维持从左到右的依赖约束,以此保障蛋白质序列的全局连贯性与生成质量;而在每个数据块内部,则通过扩散过程实现迭代去噪,使模型能够同步感知局部关键功能区域的前后文约束。这种设计巧妙地兼顾了自回归模型的高质量逻辑与扩散模型的并行高效优势。
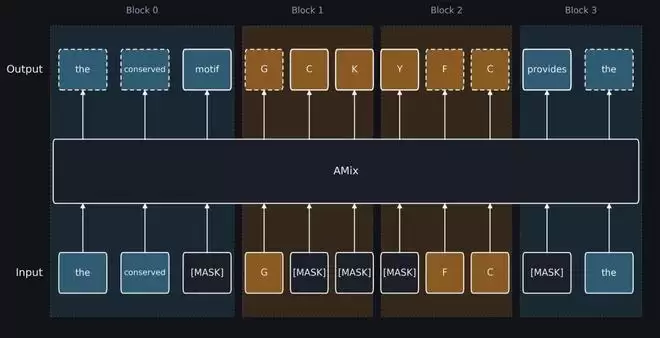
书生amix采用块级扩散(block-wise diffusion)范式,融合块间全局连贯性与块内双向感知,符合长序列的宏观逻辑与关键位点的微观约束。图中蓝色表示文本模态,橙色表示蛋白模态,虚线表示不参与训练损失计算
当前蛋白质模型评测领域存在一个关键痛点:许多模型在划分训练集与测试集时,没有进行严格的同源去重处理,这导致了蛋白质序列信息的泄露。问题在于,模型在测试时可能会依赖训练集中同源序列的信息进行推理,而非真正掌握决定蛋白质功能的底层理化规律,这使得评测结果的公正性与可信度大打折扣。
为了解决这一问题,上海AI实验室构建了ProteinArena评测集。它严格依据时间戳与同源性标准来划分各类蛋白质任务的训练集与测试集,从而有效规避了高同源蛋白质引发的信息泄露问题,为蛋白质模型评测树立了公平、严谨的新标准。
ProteinArena主要划分为原生蛋白理解与原生蛋白设计两大类任务,全面覆盖了蛋白质研究的核心需求:
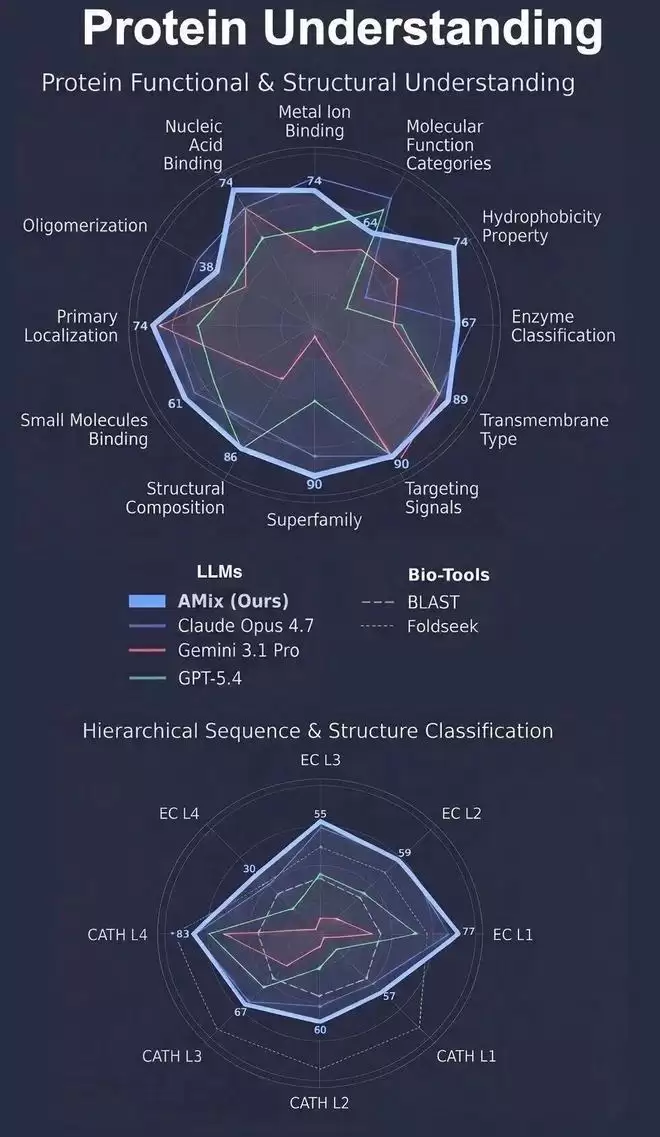
原生蛋白理解:基于2025年后Swiss-Prot数据库新收录、且与训练集同源性低于30%的蛋白质,构建了一个包含18个细分评测任务的体系。它涵盖了16类常见蛋白质知识任务、481个样本的通用蛋白质问答,以及细粒度的EC四级酶功能分类与CATH四级结构层级分类。在通用问答任务中以准确率为核心指标;而在EC与CATH任务中,除了引入通用大模型,还纳入了ESM2、ESM3等蛋白质专用模型,以及Foldseek、BLAST等经典生物信息学工具,进行多维度对比。
原生蛋白设计:参考PDFBench评测标准,采用2025年Swiss-Prot数据库新收录的审核通过蛋白质功能关键词,设置了从头设计任务,以测试模型能否依据InterPro数据库的功能描述,直接生成符合约束条件的蛋白质序列。评测重点关注两个核心指标:pLDDT(折叠可信度,数值越高表明蛋白质结构越稳定)和Function Recovery(功能恢复率,数值越高表明设计蛋白质与预期功能的契合度越高)。
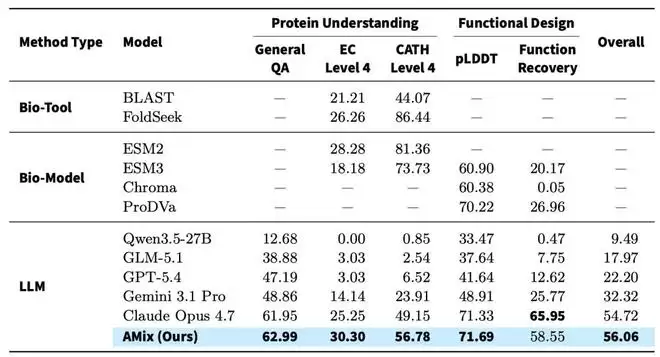
实验结果显示,在低同源蛋白质理解(general qa, ec/cath 分类)和功能设计评估中展现出卓越的性能,书生amix整体得分 (56.06) 优于现有的生物学专用模型及各大主流大语言模型
基于ProteinArena这个公平的“竞技场”,“书生·Amix”与前沿大模型、蛋白质领域专用模型以及经典工具展开了同台较量。评测结果清晰地显示:
首先,“书生·Amix”首次展现出跨越蛋白原生理解与设计模态鸿沟的基座级能力。在覆盖16类核心任务的通用蛋白质问答上,其准确率超越了Claude Opus 4.7,位居榜单第一。
其次,在最具挑战性的细粒度酶功能分类EC L4任务中,“书生·Amix”的表现领先于通用大模型、专用序列模型ESM2、ESM3以及经典检索标杆BLAST。这证明其能力已经超越了传统的序列比对与记忆范式,能够捕捉到低同源蛋白背后更深层的进化规律。
最后,在蛋白质从头设计任务中,“书生·Amix”实现了折叠可信度与功能恢复率的双重优化。它成功打破了文本与蛋白质之间的跨模态推断瓶颈,通过构建统一的表示空间实现了模态的自然对齐。这无疑为整个蛋白质研究领域的技术创新与产业应用,提供了一个堪称“革命性”的强大工具。
来源:上海人工智能实验室
编辑:朱文莹
上观号作者:上海科技
菜鸟下载发布此文仅为传递信息,不代表菜鸟下载认同其观点或证实其描述。